
The Harmony of Iron Regulating Genes and Genes of Antioxidant Enzymes in Thalassaemia Patients
Abstract:
The purpose of this work was to
investigate the expression of iron-regulating genes and their relationship to
enzymatic antioxidant genes in thalassaemic patients, as well as to detect
distinct forms of thalassaemia using an RT-PCR technique using a presence/absence
protocol. This study included 50 patients who were admitted to Mosul's Al-Hadba'a
Hospital. Ten healthy subjects and forty thalassaemic patients were aged eight
to seventeen years. Freshly blood samples were collected using EDTA for
molecular processes, as gene expression and genomic identification of
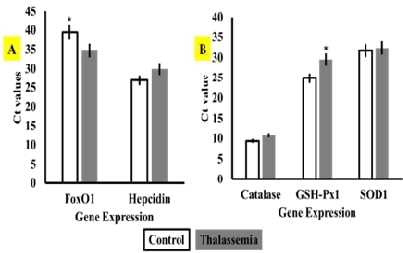
thalassaemia types. The outcomes indicated that the foxO1 gene was
significantly upregulated in thalassaemic patients compared to healthy
participants, but hepcidin expression was non-significantly downregulated.
Similarly, the enzymatic antioxidant gene GSH-Px1 demonstrated significant
downstream regulation expression in thalassaemic participants compared to healthy
ones, even though the expression of SOD1 and CAT antioxidant enzyme genes did
not differ across investigated patients. Catalase expression is associated
inversely with foxO1 expression. The presence/absence approach showed that 8.4%
and 5.1% of individuals had positive α-thalassaemia based on α1 and α2
mutations in their gDNA samples, respectively, compared to healthy patients.
Furthermore, 33.6%, 36.5%, and 16.4% of blood samples with an unknown type of
thalassaemia tested positive for ß-thalassaemia because their gDNA samples had
codon8/9, codon 41/42, and IVS-I-5 mutations, respectively. Our findings
suggest that the RT-PCR approach is the most effective for studying gene
expression and molecular identification of thalassaemia types.
References:
[1].
Britton,
R. S., Leicester, K. L., Bacon, B. R., 2002, Iron toxicity and chelation
therapy. International journal of hematology, 76, 219-228, doi: 10.1007/bf02982791.
[2].
Papanikolaou,
G., Tzilianos, M., Christakis, J. I., Bogdanos, D., Tsimirika, K., MacFarlane,
J., Nemeth, E., 2005, Hepcidin in iron overload disorders. Blood, 105(10),
4103-4105, doi: 10.1182/BLOOD-2004-12-4844.
[3].
Barisani,
D., Pelucchi, S., Mariani, R., Galimberti, S., Trombini, P., Fumagalli, D.,
Piperno, A., 2008, Hepcidin and iron-related gene expression in subjects with
Dysmetabolic Hepatic Iron Overload. Journal of hepatology, 49(1),
123-133, doi: 10.1016/J.JHEP.2008.03.011.
[4].
Latour,
C., Kautz, L., Besson‐Fournier, C., Island, M. L., Canonne‐Hergaux, F., Loréal,
O., Roth, M. P., 2014, Testosterone perturbs systemic iron balance through
activation of epidermal growth factor receptor signaling in the liver and
repression of hepcidin. Hepatology, 59(2), 683-694, doi:
10.1002/hep.26648.
[5].
Roth,
M. P., Meynard, D., Coppin, H., 2019, Regulators of hepcidin expression. Vitamins
and hormones, 110, 101-129, doi: 10.1016/BS.VH.2019.01.005.
[6].
Kassab-Chekir,
A., Laradi, S., Ferchichi, S., Khelil, A. H., Feki, M., Amri, F., Miled, A.,
2003, Oxidant, antioxidant status and metabolic data in patients with
beta-thalassemia. Clinica Chimica Acta, 338(1-2), 79-86, doi:
10.1016/j.cccn.2003.07.010.
[7].
Meral,
A., Tuncel, P., Sürmen-Gür, E., Özbek, R., Öztürk, E., GÜnay, Ü., 2000, Lipid
peroxidation and antioxidant status in ß-thalassemia. Pediatric hematology
and oncology, 17(8), 687-693, doi: 10.1080/08880010050211402.
[8].
Kattamis,
C., Lazaropoulou, C., Delaporta, P., Apostolakou, F., Kattamis, A., Papassotiriou,
I., 2011, Disturbances of biomarkers of iron and oxidant-antioxidant
homeostasis in patients with beta-thalassemia intermedia. Pediatric
endocrinology reviews: PER, 8, 256-262.
[9].
Nemeth,
E., Ganz, T., 2023, Hepcidin and iron in health and disease. Annual review
of medicine, 74(1), 261-277. https://doi.org/10.1146/annurev-med-043021-032816.
[10].
Dolai,
T. K., Nataraj, K. S., Sinha, N., Mishra, S., Bhattacharya, M., Ghosh, M. K.,
2012, Prevalance of iron deficiency in thalassemia minor: a study from tertiary
hospital. Indian Journal of Hematology and Blood Transfusion, 28, 7-9,
doi: 10.1007/S12288-011-0088-9/METRICS.
[11].
Rahman,
M., Irshadullah, N. M., Ahmed, M., Kabir, A. L., Begum, M., Mostafa, A. G.,
Khan, A. H., 2014, Prevalence of iron deficiency in thalassemia trait: a study
in BSMMU, Dhaka. Bangladesh Journal of Medicine, 25(1), 13-16, doi: 10.3329/BJMED.V25I1.25072.
[12].
Tantiworawit,
A., Khemakapasiddhi, S., Rattanathammethee, T., Hantrakool, S.,
Chai-Adisaksopha, C., Rattarittamrong, E., Fanhchaksai, K., 2021, Correlation
of hepcidin and serum ferritin levels in thalassemia patients at Chiang Mai
University Hospital. Bioscience Reports, 41(2), BSR20203352, doi:
10.1042/BSR20203352.
[13].
Haghpanah,
S., Esmaeilzadeh, M., Honar, N., Hassani, F., Dehbozorgian, J., Rezaei, N., Karimi,
M., 2015, Relationship between serum hepcidin and ferritin levels in patients
with thalassemia major and intermedia in Southern Iran. Iranian Red Crescent
Medical Journal, 17(7), doi: 10.5812/ircmj.17(5)2015.28343.
[14].
Susanah,
S., Rakhmilla, L. E., Ghozali, M., Trisaputra, J. O., Moestopo, O., Sribudiani,
Y., Maskoen, A. M., 2021, Iron Status in Newly Diagnosed β‐Thalassemia Major:
High Rate of Iron Status due to Erythropoiesis Drive. BioMed Research
International, 2021(1), 5560319, doi: 10.1155/2021/5560319.
[15].
Eshagh
Hossaini, S. K., Haeri, M. R., 2019, Association between serum levels of
hepcidin and ferritin in patients with thalassemia major and intermedia, the
role of iron chelator. Journal of Hematopathology, 12, 143-147, doi:
10.1007/S12308-019-00363-X/FIGURES/4.
[16].
Lim,
W. F., Muniandi, L., George, E., Sathar, J., Teh, L. K., Lai, M. I., 2015, HbF
in HbE/β-thalassemia: A clinical and laboratory correlation. Hematology,
20(6), 349-353, doi: 10.1179/1607845414Y.0000000203.
[17].
Voskou,
S., Aslan, M., Fanis, P., Phylactides, M., Kleanthous, M., 2015, Oxidative
stress in β-thalassaemia and sickle cell disease. Redox biology, 6,
226-239, doi: 10.1016/J.REDOX.2015.07.018.
[18].
Rifkind,
J. M., Mohanty, J. G., Nagababu, E., 2015, The pathophysiology of extracellular
hemoglobin associated with enhanced oxidative reactions. Frontiers in
physiology, 5, 500, doi: 10.3389/FPHYS.2014.00500/BIBTEX.
[19].
Johnson,
R. M., Ho, Y. S., Yu, D. Y., Kuypers, F. A., Ravindranath, Y., Goyette, G. W.,
2010, The effects of disruption of genes for peroxiredoxin-2, glutathione
peroxidase-1, and catalase on erythrocyte oxidative metabolism. Free Radical
Biology and Medicine, 48(4), 519-525, doi:
10.1016/J.FREERADBIOMED.2009.11.021.
[20].
Nagababu,
E., Mohanty, J. G., Friedman, J. S., Rifkind, J. M., 2013, Role of
peroxiredoxin-2 in protecting RBCs from hydrogen peroxide-induced oxidative
stress. Free radical research, 47(3), 164-171, doi:
10.3109/10715762.2012.756138.
[21].
Homma,
T., Okano, S., Lee, J., Ito, J., Otsuki, N., Kurahashi, T., Fujii, J., 2015,
SOD1 deficiency induces the systemic hyperoxidation of peroxiredoxin in the
mouse. Biochemical and biophysical research communications, 463(4),
1040-1046, doi: 10.1016/J.BBRC.2015.06.055.
[22].
Al-Allawi,
N., Al Allawi, S., Jalal, S. D., 2021, Genetic epidemiology of
hemoglobinopathies among Iraqi Kurds. Journal of Community Genetics,
12(1), 5-14, doi: 10.1007/s12687-020-00495-z.
[23].
Suwannakhon,
N., Pangeson, T., Seeratanachot, T., Mahingsa, K., Pingyod, A., Bumrungpakdee,
W., Sanguansermsri, T., 2019, Noninvasive prenatal screening test for compound
heterozygous beta thalassemia using an amplification refractory mutation system
real-time polymerase chain reaction technique. Hematology Reports, 11(3),
doi: 10.4081/hr.2019.8124.
[24].
Al-Allawi,
N. A., Puehringer, H., Raheem, R. A., Oberkanins, C., 2015, Genetic modifiers
in β-thalassemia intermedia: a study on 102 Iraqi Arab patients. Genetic
testing and molecular biomarkers, 19(5), 242-247, doi:
10.1089/gtmb.2014.0310.
[25].
Kho,
S. L., Chua, K. H., George, E., Tan, J.A.M. A., 2015, A novel gap-PCR with high
resolution melting analysis for the detection of α-thalassaemia Southeast Asian
and Filipino β0-thalassaemia deletion. Scientific Reports, 5, doi:
10.1038/srep13937.
[26].
Lazarte,
S. S., Mónaco, M. E., Haro, A. C., Jiménez, C. L., Ledesma Achem, M. E., Issé,
B. A., 2014, Molecular characterization and phenotypical study of β-thalassemia
in Tucumán, Argentina. Hemoglobin, 38(6), 394-401, doi:
10.3109/03630269.2014.968784.
[27].
Saud,
A. M., 2012, Molecular and biochemical study on β-thalassemia patients in Iraq.
University of Baghdad.pdf
[28].
Marashi,
S. J., Eshkoor, S. A., saed Mirinargesi, M., Sarookhani, M. R., Rahmat, A. B.,
Ismail, P. B., 2012, Detection of eight common [beta]-globin gene mutation in
thalassemia major patients using real time polymerase chain reaction (PCR)-high
resolution melting and EvaGreen (TM) dye. African Journal of Biotechnology,
11(2), 448, doi: 10.5897/ajb10.1167.
[29].
Al-Allawi,
N. A., Badi, A. I., Imanian, H., Nikzat, N., Jubrael, J. M., Najmabadi, H., 2009,
Molecular characterization of α-thalassemia in the Dohuk region of Iraq. Hemoglobin,
33(1), 37-44, doi: 10.1080/03630260802626053.
[30].
Pornprasert,
S., Phusua, A., Suanta, S., Saetung, R., Sanguansermsri, T., 2008, Detection of
alpha‐thalassemia‐1 Southeast Asian type using real‐time gap‐PCR with SYBR
Green1 and high resolution melting analysis. European journal of
haematology, 80(6), 510-514, doi: 10.1111/j.1600-0609.2008.01055.x.
[31].
Gaafar,
T. M., ELBeshlawy, A. M., Aziz, M. I., Abdelrazik, H. N., 2006, Rapid screening
of β-Globin gene mutations by Real-Time PCR in Egyptian thalassemic children. African
Journal of Health Sciences, 13(3), 70-77, doi: 10.4314/ajhs.v13i3.30839.
[32].
Al-Allawi,
N. A., Jubrael, J. M., Hughson, M., 2006, Molecular characterization of
β-thalassemia in the Dohuk region of Iraq. Hemoglobin, 30(4), 479-486,
doi: 10.1080/03630260600868097.
[33].
Sun,
C. F., Lee, C. H., Cheng, S. W., Lin, M. H., Wu, T. L., Tsao, K. C., Chu, D. C.,
2001, Real‐time quantitative PCR analysis for α‐thalassemia‐1 of Southeast
Asian type deletion in Taiwan. Clinical genetics, 60(4), 305-309, doi:
10.1034/j.1399-0004.2001.600409.x.
[34].
Oron‐Karni,
V., Filon, D., Oppenheim, A., Rund, D., 1998, Rapid detection of the common
Mediterranean α‐globin deletions/rearrangements using PCR. American journal
of hematology, 58(4), 306-310, doi:
10.1002/(SICI)1096-8652(199808)58:4<306::AID-AJH10>3.0.CO;2-5.
[35].
Chang,
J. G., Liu, H. J., Huang, J. M., Yang, T. Y., Chang, C. P., 1997, Multiplex
mutagenically separated PCR: diagnosis of β-thalassemia and hemoglobin
variants. Biotechniques, 22(3), 520-527, doi: 10.2144/97223rr03.
[36].
Rosnah,
B., Rosline, H., Zaidah, A. W., Noor Haslina, M. N., Marini, R., Shafini, M. Y.,
Nurul Ain, F. A., 2012, Detection of Common Deletional Alpha‐Thalassemia
Spectrum by Molecular Technique in Kelantan, Northeastern Malaysia. International
Scholarly Research Notices, 2012(1), 462969, doi: 10.5402/2012/462969.
[37].
Farra,
C., Badra, R., Fares, F., Muwakkit, S., Dbaibo, G., Dabbous, I., Abboud, M. R.,
2015, Alpha thalassemia allelic frequency in Lebanon. Pediatric Blood &
Cancer, 62(1), 120-122, doi: 10.1002/PBC.25242.
[38].
Hadavi,
V., Taromchi, A. H., Malekpour, M., Gholami, B., Law, H. Y., Almadani, N.,
Najmabadi, H., 2007, Elucidating the spectrum of α-thalassemia mutations in
Iran. haematologica, 92(7), 992-993, doi: 10.3324/HAEMATOL.10658.
[39].
Adekile,
A., Sukumaran, J., Thomas, D., D’Souza, T., Haider, M., 2020, Alpha thalassemia
genotypes in Kuwait. BMC Medical Genetics, 21, 1-5, doi:
10.1186/S12881-020-01105-Y/TABLES/4.
[40].
Baysal,
E., 2011, α-Thalassemia syndromes in the United Arab Emirates. Hemoglobin,
35(5-6), 574-580, doi: 10.3109/03630269.2011.634698.
