
Enhanced HACBLalign Method using Transitional Pattern Search and Pre-Trained Classification Model for Protein Remote Homology Detection and Fold Recognition
Abstract:
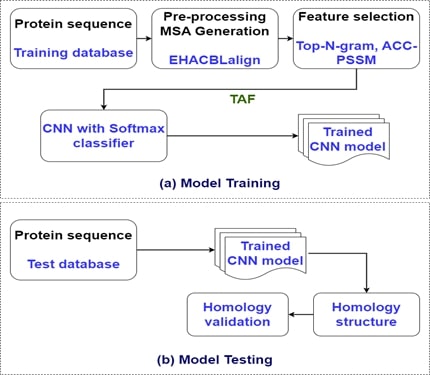
One
of the most important tasks to predict the structure of proteins is Protein
Remote Homology Detection and Fold Recognition. To do this, a Hierarchical
Attention-based Convolutional Neural Network with Bidirectional Long Short-Term
Memory called the HACBLalign algorithm was proposed by the authors, which
performs Multiple Sequence Alignments (MSAs), extracts features, and recognizes
protein homologies. But, when the quantity of Protein Sequences (PSs)
increases, the number of times the decision-making system runs also increases. To
avoid this issue, this article proposes an Enhanced HACBLalign (EHACBLalign) method
using Transitional Pattern Search (TPS) and pre-trained classification for Protein
Remote Homology Detection and Fold Recognition. During the alignment stage, the
intermediate sequences such as Hit Regions (HRs) are identified by the TPS. Then,
the HRs are extended in middle layers and utilized as a query in all TPS
iterations. Besides, the HACBLalign algorithm is applied in all intermediate
layers for generating pairwise alignments. Moreover, each pairwise alignment
between intermediate sequences is merged to get the final alignment. Further,
various characteristics are obtained from the chosen alignment and learned by
the pre-trained Convolutional Neural Network (CNN) with a softmax function for recognizing
protein remote homologies precisely. This enhances the performance of the decision-making
system for large-scale PS databases. Finally, the test outcomes exhibit that
the EHACBLalign realizes a 94.6%, 94.1%, and 93.4% accuracy on SCOP 1.53, SCOP
1.67, and superfamily corpora, respectively in Protein Remote Homology
Detection and Fold Recognition.
References:
[1]. Lv, Z., Ao, C., and Zou, Q., 2019,
Protein function prediction: from traditional classifier to deep learning, Proteomics, 19(14), 1-5.
[2]. Jing, X., Dong, Q., Hong, D., and
Lu, R., 2019, Amino acid encoding methods for protein sequences: A comprehensive
review and assessment, IEEE/ACM
Transactions on Computational Biology and Bioinformatics, 17(6), 1918-1931.
[3]. Rajapaksa, S, Sumanaweera, D, Lesk,
A, M, Allison, L, Stuckey, P, J, Garcia de la Banda, M, and Konagurthu, A, S.,
2022, On the reliability and the limits of inference of amino acid sequence
alignments, Bioinformatics, 38(Supplement_1), i255-i263.
[4]. Peyravi, F, Latif, A, and
Moshtaghioun, S. M., 2019, Protein tertiary structure prediction using hidden
Markov model based on lattice, Journal
of Bioinformatics and Computational Biology, 17(02), 1-18.
[5]. Wilburn, G. W., and Eddy, S. R., 2020.
Remote homology search with hidden Potts model, PLOS Computational Biology, 16(11), 1-22.
[6]. Chen, J., Guo, M., Wang, X., and
Liu, B., 2018, A comprehensive review and comparison of different computational
methods for protein remote homology detection, Briefings in Bioinformatics, 19(2), 231-244.
[7]. Li, C, C., and Liu, B., 2020,
MotifCNN-fold: protein fold recognition based on fold-specific features
extracted by motif-based convolutional neural networks, Briefings in Bioinformatics, 21(6), 2133-2141.
[8]. Wu, Z., Liao, Q., and Liu, B., 2020,
A comprehensive review and evaluation of computational methods for identifying
protein complexes from protein–protein interaction networks, Briefings in Bioinformatics, 21(5), 1531-1548.
[9]. Liu, B., Chen, J., Guo, M., and
Wang, X., 2017, Protein remote homology detection and fold recognition based on
sequence-order frequency matrix, IEEE/ACM
Transactions on Computational Biology and Bioinformatics, 16(1), 292-300.
[10]. Guo, Y., Yan, K., Wu, H., and Liu,
B., 2020, ReFold-MAP: Protein remote homology detection and fold recognition
based on features extracted from profiles, Analytical Biochemistry, 611, 1-8.
[11]. Gopinath, K., and Rajendran, G., 2023,
HACBLalign: A Hierarchical Attention-based deep learning for protein remote
homology and fold identification, Journal of Theoretical and Applied
Information Technology, 14(101), 5578 – 5588.
[12]. Makigaki, S, and Ishida, T., 2020,
Sequence alignment using machine learning for accurate template-based protein
structure prediction, Bioinformatics, 36(1), 104-111.
[13]. Zhang, C., Zheng, W., Mortuza, S. M.,
Li, Y., and Zhang, Y., 2020, Deep MSA: Constructing deep multiple sequence
alignment to improve contact prediction and fold-recognition for
distant-homology proteins, Bioinformatics, 36(7), 2105-2112.
[14]. Senior, A. W., Evans, R., Jumper, J.,
Kirkpatrick, J., Sifre, L., Green, T., and Hassabis, D., 2020, Improved protein
structure prediction using potentials from deep learning, Nature, 577(7792), 706-710.
[15]. Wu, F., and Xu, J., 2021, Deep
template-based protein structure prediction, PLoS Computational Biology, 17(5), 1-18.
[16]. Wu, T., Guo, Z., Hou, J., and
Cheng, J., 2021, DeepDist: Real-value inter-residue distance prediction with
deep residual convolutional network, BMC
Bioinformatics, 22(1),
1-17.
[17]. Hakala, K., Kaewphan, S., Bjorne, J.,
Mehryary, F., Moen, H., Tolvanen, M., and Ginter, F., 2022, Neural network and
random forest models in protein function prediction. IEEE/ACM Transactions on Computational Biology and Bioinformatics,
19(3), 1772-1781.
[18]. Liu, J., Wu, T., Guo, Z., Hou, J., and
Cheng, J., 2022, Improving protein tertiary structure prediction by deep
learning and distance prediction in CASP14. Proteins: Structure, Function, and Bioinformatics, 90(1), 58-72.
[19]. Zhang, C., and Pyle, A, M., 2022, A
unified approach to sequential and non-sequential structure alignment of
proteins, RNAs and DNAs, Iscience,
25(10), 1-13.
[20]. Rangwala, H, and Karypis, G., 2005,
Profile-based direct kernels for remote homology detection and fold
recognition, Bioinformatics, 21(23), 4239-4247.
[21]. Håndstad, T., Hestnes, A. J., and
Sætrom, P., 2007, Motif kernel generated by genetic programming improves remote
homology and fold detection, BMC
Bioinformatics, 8(1), 1-16.
[22]. Andreeva, A., Kulesha, E., Gough, J.,
and Murzin, A, G., 2020, The SCOP database in 2020: expanded classification of
representative family and superfamily domains of known protein
structures. Nucleic Acids Research, 48(D1), D376-D382.
[23]. Devlin, J., Chang, M. W., Lee, K., and
Toutanova, K., 2018, Bert: Pre-training of deep bidirectional transformers for
language understanding, arXiv preprint arXiv:1810.04805.
